CLINUVEL Communiqué 1
| Melbourne, Australia, 27 January 2022 |
ASX: XETRA-DAX: Level 1 ADR: |
CUV UR9 CLVLY |
Dear Shareholders, Friends
Introduction
(Mr Malcolm Bull, Head of Australian Operations & Investor Relations)
Happy New Year to all our stakeholders for 2022, a year in which CLINUVEL anticipates advances in our strategic initiatives across all Divisions of our business. As we enter the third year of the COVID–19 pandemic, we take encouragement from the world’s progress to develop and administer vaccines. We empathise with all those affected by the pandemic and admire and appreciate the dedication of healthcare workers around the world. Whilst we continue to manage impacts on our business and monitor the distribution of booster vaccines to combat the Delta and Omicron variants, the CLINUVEL team is fortified to progress the growth of commercial operations and the expansion of the clinical program.
CLINUVEL‘s financial management provides the Company the ability to manage externalities, such as the pandemic. Our objectives have therefore not changed and CLINUVEL continues to make headway, notwithstanding the prevailing operating environment, towards building an independent group of companies.
This first news communiqué of 2022 provides an update on:
- Commercial operations;
- Peer reviews of EPP patient experience with SCENESSE®;
- Clinical programs; and
- Communications and investor relations.
Whilst we continue to manage impacts on our business and monitor the distribution of booster vaccines to combat the Delta and Omicron variants, the CLINUVEL team is fortified to progress the growth of commercial operations and the expansion of the clinical program
Treatment for Erythropoietic Protoporphyria (EPP) Patients in the USA
(Dr Linda Teng, Director North American Operations)
Specialty Centers and Insurers
Over 40 Specialty EPP Centers have been trained and accredited across the US. These centers are strategically situated in proximity to EPP patients’ residences. The US team continues to identify and train centers in new locations based on the cluster of known EPP patients. Since mid 2021 we have seen an increase from 60 to over 100 commercial and state insurers, which have now published clinical policy documents for SCENESSE® and are covering the SCENESSE® treatment under Prior Authorization (PA). With these clinical policies in place, patients with confirmed EPP diagnoses are receiving PA approvals and coverage of the medication.
In addition, the Medicare program has begun covering SCENESSE® for a small group of EPP patients. Medicare, the federal health insurance program administered by the Centers for Medicare & Medicaid Services (CMS), provides health insurance for Americans who are 65 or older, certain younger people with disabilities, and Americans with End-Stage Renal Disease.
The SCENESSE® Savings Program was established for eligible EPP patients under private and/or commercial insurance plans. This program has been successfully implemented and has provided financial assistance to eligible EPP patients since the start of market access in 2020.
Since mid-2021 we have seen an increase from 60 to over 100 commercial and state insurers, which have now published clinical policy documents for SCENESSE® and are covering the SCENESSE® treatment under Prior Authorization (PA).
Treatment Codes, Implementation and Effects
Treatment codes for the billing of SCENESSE® medication and procedure on insurance claims have now been fully established. The level II HCPCS medication J-code (J7352) for SCENESSE® as “afamelanotide implant, 1 mg” was assigned by the HCPCS (Healthcare Common Procedure Coding System) in January 2021. The administration procedural code CPT 11981 “Insertion, drug delivery implant (i.e., bioresorbable, biodegradable, nonbiodegradable)” was approved and revised by the AMA (American Medical Association) in 2021 with an effective date of January 2022.
In most cases, coverage of SCENESSE® (J7352) continues to require PA approval while the updated procedure code for SCENESSE® administration (CPT 11981) no longer requires PA approval. This change will decrease the Centers’ administrative time. Insurers are appropriately reimbursing SCENESSE® according to the J7352 contracted rate between individual Centers and insurers. In implementing the changes in code registration, we assist or actively manage the discrepancies, errors and delays incurred for patients to receive treatment, but overall, a first assessment is that the new billing code has improved the efficiency of the billing process for insurers and the Centers alike.
Patient Demand and Experiences
In reality, last year (2021) was the first complete calendar year of SCENESSE® treatment and it was observed that a majority of EPP patients continued to receive treatment throughout the winter season. While CLINUVEL reports following the Australian financial year (1 July to 30 June), we also monitor the calendar year performance for regulatory purposes. As seen in previous years, higher demand for treatment began around the spring and summer seasons. We will await whether the same trend is to be seen this coming year, while our team will continue to refer EPP patients to Specialty Centers and assist them with PA approvals. We witnessed that in the past few weeks there has been an increase in activity on PA submissions and/or renewals in preparation for the spring season.
Throughout the past year, our clinical and distribution teams have received positive feedback and improved quality of life anecdotes post-SCENESSE® treatment. These real-life experiences provide us with valuable insights into how patients benefit and are able to lead lives previously thought unimaginable. With increased vigour and new team members, we look forward to assisting Specialty Centers and patients to ensure that they continue to receive exceptional care and improved quality of life from SCENESSE® treatment.
We have a specific plan we stick to when it comes to long-term clinical care, and the fruits are paying off in Europe and the US, providing the supervisory bodies much confidence that we actually do look after our patients.
Treatment for EPP Patients in Europe
(Mr Lachlan Hay, Director of Global Operations)
EPP patients persist in seeking treatment with SCENESSE® (afamelanotide 16mg) across Europe, despite the ongoing restrictions posed by the COVID–19 pandemic. Based on initial feedback we expect some European EPP Expert Centres to continue to operate at limited capacity during 2022, as resources are diverted to critical patient care and catching up with the backlog of elective care. However, the EPP Expert Centres are aware of the advantages of offering continuous care for EPP patients rather than taking them off treatment (see Dr Bilbao’s commentary below). EPP remains a complex disease requiring multidisciplinary care.
Continuation of SCENESSE® in Germany
As announced earlier this week, a most important milestone has been achieved by our team securing a second agreement with the German National Association of Statutory Health Insurance Funds (GKV-SV). This agreement reassures German EPP patients access to SCENESSE®. In preparation of these lengthy but necessary negotiations, our team analysed distribution and patients’ data, revisited clinical trial data and the treatment response rate. A team of experts, counsel, and clinical and commercial members concluded that CLINUVEL’s position and approach to negotiate a fair and equitable price was justified. Bolstered with confidence that CLINUVEL had kept its promise towards German (but also other) national insurers, the discussions with the Federal Joint Committee (G-BA) and GKV-SV proceeded, behind closed doors.
Two public meetings were held when our teams uncovered an unfair attempt by the negotiators to present other unfounded data, but eventually during the negotiations it was demonstrated from CLINUVEL’s audited data that our position was rock solid and based on facts and data. I have called this positive outcome in CLINUVEL’s public announcement (ASX: 24 January 2022) the direct result of our strategy and equitable approach across Europe.
There are many EPP patients and families in Germany who have received ongoing care and treatment at EPP Expert Centres since the commencement of our commercial program in 2016, with a number having previously participated in our clinical trial and compassionate access programs. All these patients provide valuable real-life data, complementing the clinical trial data already produced.
…an important milestone has been achieved by our team by securing a second agreement with the German National Association of Statutory Health Insurance Funds (GKV-SV). This agreement reassures German EPP patients access to SCENESSE®
The reimbursement process in Germany is legally prescribed in the German Social Book V (Sozialgesetzbuch V or SGB V) and involves interactions with and between three distinct but overlapping entities:
- the G-BA;
- the Institute for Quality and Efficiency in Healthcare (iQWiG); and
- the GKV-SV
A further Framework Agreement – defined under Section 130b of the SGB V – mandates the processes and interactions between pharmaceutical Marketing Authorisation Holders (MAHs) and the GKV-SV to arrive at agreements on reimbursement, which affects the ability of around 90% of the German population to access treatment. (The other 10% of the population is privately insured through members of the Private Krankenversicherung, PKV, who are an observer to the GKV process.) Eventually, the Ministry of Health will also be involved in assessing market access of novel therapies.
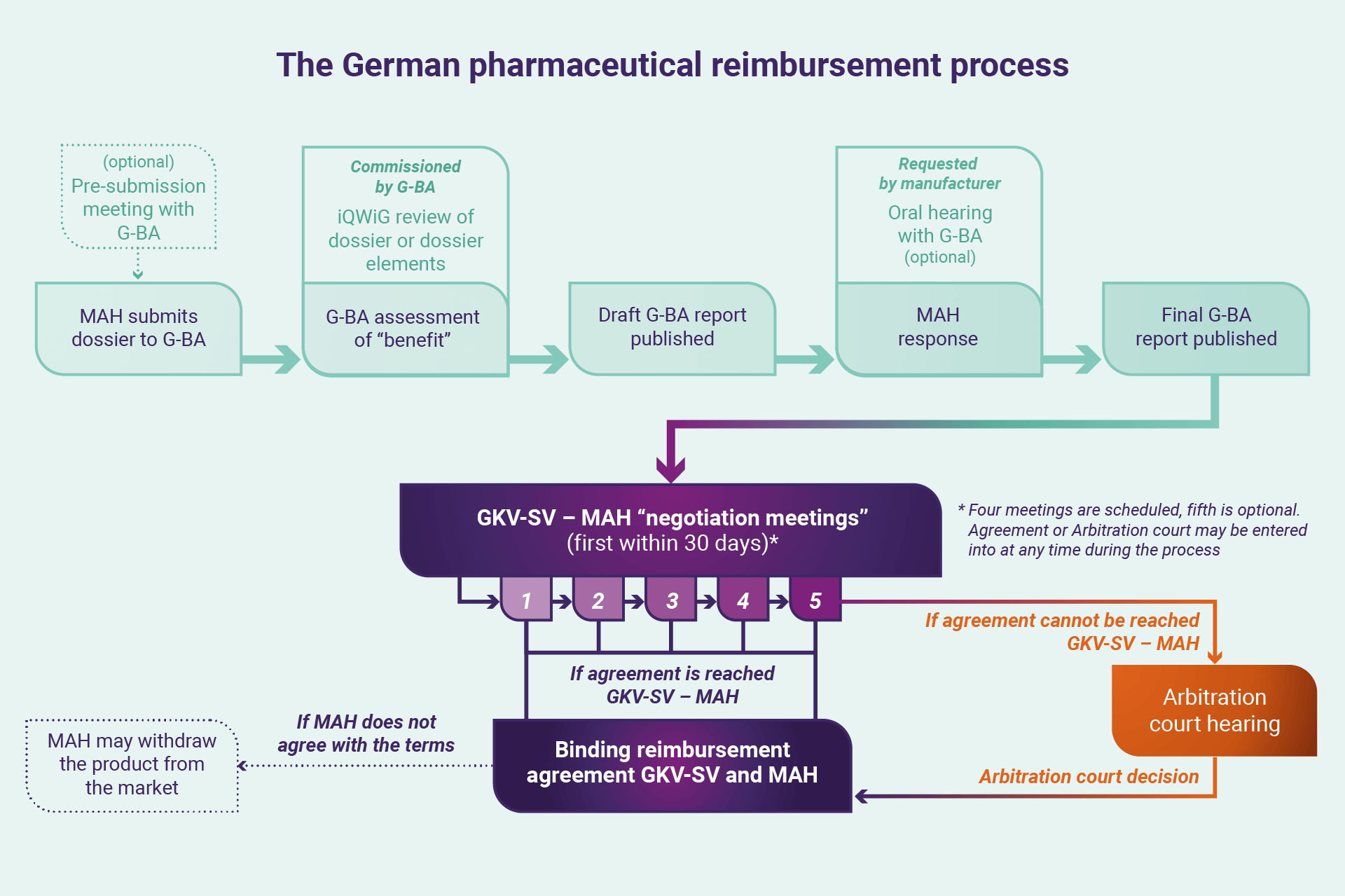
From first interaction to final agreement, discussions and price negotiations can take 18–24 months, a pathway all companies must follow to enable or continue market access for therapies in Germany. For the first 12 months after the launch of a product in Germany, an MAH can set the market price of a product freely but may be subject to heavy payment clawbacks in the subsequent periods while the GKV-SV process is finalised.
The first interactions and discussions in the German reimbursement process focus on the “benefit” of a medication, irrespective of the benefit assessments already conducted by the European Medicines Agency (EMA).
The G-BA Approach to “benefit”
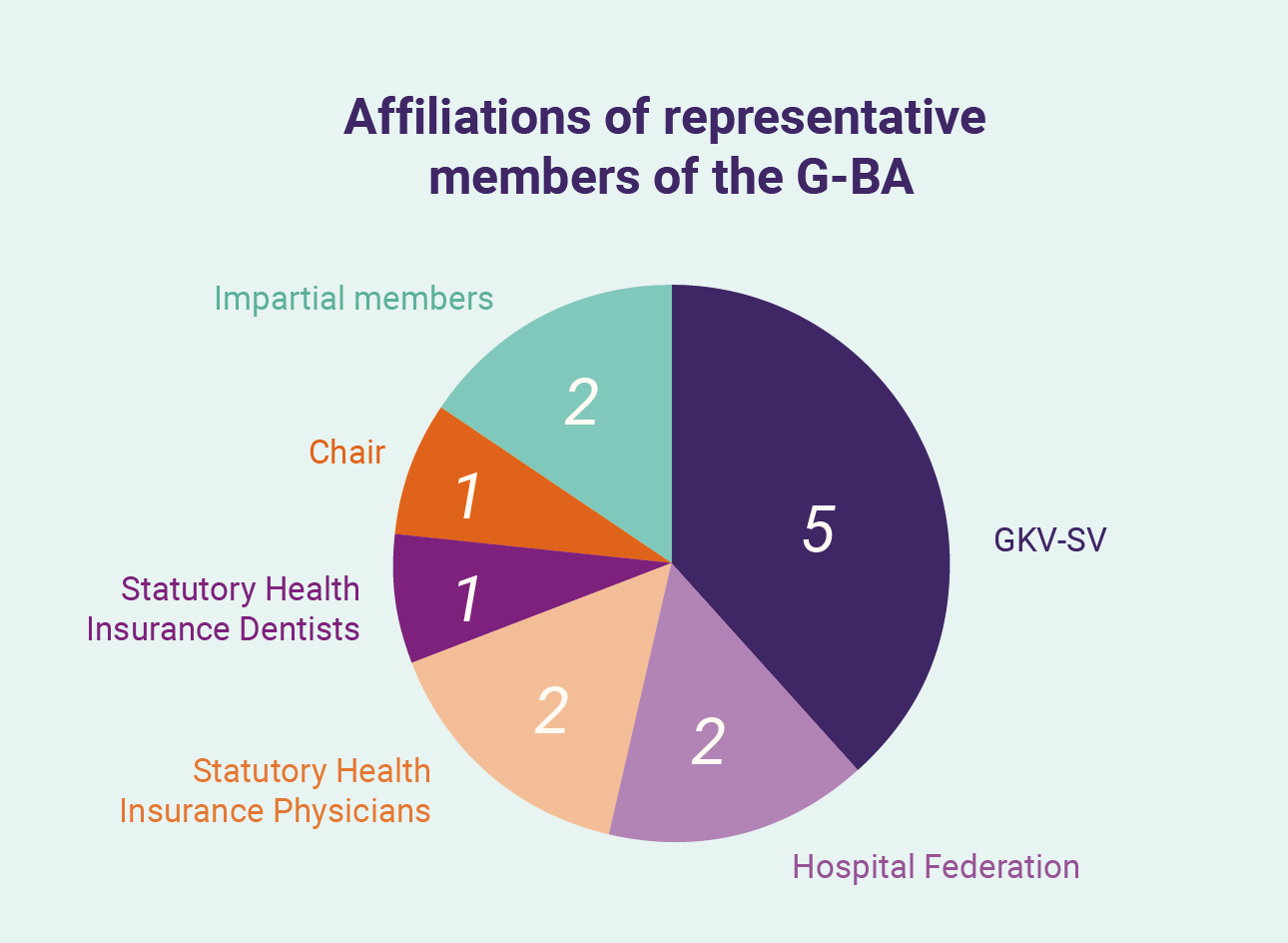
The first interactions and discussions in the German reimbursement process focus on the “benefit” of a medication, irrespective of the benefit assessments already conducted by the European Medicines Agency (EMA). A benefit assessment is made by the G-BA, a 13-member panel comprising: two representatives each of the Hospital Federation and Statutory Health Insurance Physicians; one representative of the Statutory Health Insurance Dentists; five representatives of the GKV-SV; two impartial members; and an impartial chair. Section 35a of SGB V requires this evaluation of the benefit (including against an “appropriate comparator”, as an “additional benefit” and the therapeutic significance) of a medicinal product by G-BA, with MAHs required to submit dossiers and evidence to support their claims. Companies can seek to engage G-BA prior to this submission to understand how benefit may be understood by the G-BA and refine the evidence submitted. These dossiers – running several hundred pages – cover the development and clinical trials for a product in a specific patient population, additional evidence collected (such as real-world evidence) and extensive peer-review literature. G-BA may seek further information or documents in response to the submission over subsequent months as it prepares two documents for publication: a benefit assessment report (“Nutzenbewertung”) and a supporting reasons report (“Tragende Gruende”).
For an orphan drug, the “benefit” of the product is already recognised by German legislation (unless sales are expected to exceed a defined threshold), and the role of the G-BA is primarily to quantify the benefit of the product, categorising according to low-medium-high or “unquantifiable” in the Tragende Gruende. This categorisation has a direct impact upon the later interactions with GKV-SV.
The G-BA is not limited in the evidence it can choose to accept or reject in its assessment, down to the level of individual endpoints in specific clinical studies. These are categorised according to their effects on patient mortality, morbidity, quality of life and safety. During the review, the iQWiG may be commissioned by G-BA to conduct independent assessments of a full dossier, or specific elements of a dossier according to the needs defined by G-BA, including particular evidence or endpoints, or broader information (such as the size of a patient population). The Nutzenbewertung outlines the rationale for acceptance or rejection of evidence, including each of the endpoints identified based on their operationalisation, patient relevance, and validity. Further assessments include whether evidence collected in other regions are applicable to the German situation, and whether G-BA views certain evidence or studies as biased.
Around four months from the dossier submission, a draft of the Nutzenbewertung is published and open for feedback from the MAH, who can also request an oral hearing to discuss the reports, evidence, and decisions taken by G-BA. The MAH can again present its case at the oral hearing and seek to address specific points raised by the G-BA (or iQWiG) in the reports. A final Nutzenbewertung is then prepared and published.
From G-BA to the GKV-SV
The reimbursement process then shifts from interactions with the G-BA to the GKV-SV. Under the Framework Agreement, the GKV-SV uses the Nutzenbewertung as the basis for discussing the benefit of a therapy in the negotiation process: a series of four meetings (with an optional fifth) of four hours each (in reality mostly covering many more), where representatives of the GKV-SV and MAH are asked to arrive at an agreement to enable or continue access to a therapy. Negotiation discussions – held roughly a month apart – initially focus on disease and benefit of treatment, then into pricing, reimbursement (generally focused on expected discounts), and overall national and regional budget impact. Of particular relevance are the prices charged for a therapy by a manufacturer in other countries, the level of discounting offered, and the costs of comparator therapies for the disease. In general, GKV-SV expects a discount from manufacturers to enable entry to the German market above and beyond the normally mandated seven percent. The negotiations are kept as confidential as dictated by statutory obligations.
Each of the GKV-SV meetings are held in plenary (virtually in contemporary times), chaired by the GKV-SV with minutes taken. Outside of the formal meetings, either party also may approach the other to seek an agreement, or alternatively declare that they see the differences as irreconcilable and seek intervention from a Healthcare Court of Arbitration, the decisions of which are ultimately binding on both parties. In the event a manufacturer does not accept the findings of the Court, they may ultimately withdraw their product from the German market. Therefore, an extensive list of prescriptive drugs may be withdrawn from the German market each year. If an agreement can be reached throughout the process, however, a formal contract may be signed between both parties.
A Successful Outcome
For a second time in six years, CLINUVEL’s team has come to a successful end of this process, with the 2021 reassessment by G-BA triggered by a time limit set on their initial decision in 2016. In 2017, the benefits of SCENESSE® and its fair clinical value were recognised by the Healthcare Court of Arbitration, enabling German patients the necessary treatment access. Consistent with our public statements, and evidenced by annual financial audits, the Company has implemented a transparent uniform pricing policy, not offering rebates, discounts or cashbacks which are most commonly expected in pricing discussions with state and private insurers, and often the only mechanism available to companies to enable patient access.
In our interactions since 2017, and particularly throughout 2021, German insurers have gained much confidence in CLINUVEL’s policies and come to understand how they, too, benefit from such an equitable approach. While, like all GKV-SV agreements, the details must be kept confidential, the ultimate outcome is that EPP patients in Germany can continue to receive treatment. It must also be seen as a broader victory for EPP patients across Europe, with Germany seen as one important reference country for reimbursement decisions taken.
While we are satisfied and remain humble on the GKV-SV outcome, we continue to appreciate the engagement of the GKV-SV leadership. Our team has been regularly frustrated by the way in which patient treatment and data have been misrepresented or ignored by representatives of some bodies responsible for making benefit assessments which ultimately present barriers for EPP patients to access the only treatment which has been approved for their condition. Ultimately, insurers will look at the economics and allocate funds to other treatments or where rebates are paid.
In our interactions since 2017, and particularly throughout 2021, German insurers have gained much confidence in CLINUVEL’s policies and come to understand how they, too, benefit from such an equitable approach.
SCENESSE® was approved by the EMA in 2014. In approving the product, the Agency – with the registration dossier review led by rapporteurs from the German agency BfArM – recognised the unique nature of EPP as a metabolic disorder and the challenges posed in quantifying the impact of the disease on a patient (and thus the impact of a therapy). This was summarised in the European Public Assessment Report for SCENESSE® thus:
“Under normal conditions of use, the status of current scientific knowledge, tools and instruments, does not allow for sufficient precise measurements of impact of disease and ‘visible light’ to exposed skin.”
The EMA also recognised that it would be unethical to conduct further clinical trials of afamelanotide in EPP patients, a patient population, who may not be able or willing to participate in further studies due to the complexities and risk of incurring 3rd degree burns as they suffer from absolute light intolerance. Put differently, the unique challenges of EPP require one making judgements on treatment benefit to consider approaches other than the “usual” assessment routes. As evidenced by the US Food and Drug Administration – who incorporated real world evidence into their positive benefit-risk evaluation of SCENESSE® for EPP – such approaches are, at least, becoming more widely recognised as a way forward in evaluating novel therapies. In many ways, SCENESSE® was a benchmark for the EMA and FDA to refer to for other orphan medicinal products.
Peer Reviews of Experience of EPP Patients with SCENESSE®
(Dr Pilar Bilbao, Head of Clinical Operations)
As we have progressed the post-authorisation and special access programs in Europe, our team has learnt more about EPP and its treatment under real world conditions, adding valuable insights, validating assessment tools, and – above all else – closely (often daily) monitoring the product’s safety profile. The body of data concerning the use of the product in EPP continues to grow and give us comfort as to the long-term benefit-risk profile of afamelanotide.
Two recently released peer-review articles from European expert centres illustrate this, with one providing exciting insight to a previously unreported effect in EPP patients: protection from liver injury. While our team had an inkling on the possible systemic and hepatic effects of afamelanotide 16 mg in controlled-release form, it was the Swiss group which is credited with finding evidence of hepatic protection provided during long-term use.
Throughout their lifetimes, approximately up to 20 percent of EPP patients experience some sort of liver injury, a result of the accumulation and storage of protoporphyrin IX – PPIX; the same precursor porphyrin responsible for phototoxicity – in the liver. In approximately 2 to 5 percent of EPP patients, this damage progresses to liver failure, necessitating a life-saving liver transplant and subsequent lifelong immunosuppression. Our teams have long focussed on the medical conditions arising from immune suppression, knowledge we use in other programs such as the current DNA repair trials. However, this severe EPP symptom of hepatoxicity requires that EPP patients’ liver function is assessed regularly, and that patients take precautionary measures to avoid liver damage, avoiding certain pharmaceutical products as well as alcohol.
Two recently released peer-review articles from European expert centres illustrate this, with one providing exciting insight to a previously unreported effect in EPP patients: protection from liver injury.
Liver damage can be assessed with a blood test expressing specific enzymes and proteins. Specifically, in EPP patients, PPIX levels are measured, with elevated levels suggesting the liver may be under strain to filter toxins from the blood circulation.
EPP patients in Switzerland have been receiving afamelanotide treatment across various programs since as early as 2006, with the first ever clinical trial conducted at the Triemli Hospital in Zurich (subsequently published in the New England Journal of Medicine). In a study1 conducted over two years, specific laboratory values in 38 EPP patients were assessed, concluding that treatment with afamelanotide significantly reduced the levels of both PPIX and aspartate aminotransferase (AST), an enzyme which can be produced by the liver in response to damage. The reduction was most pronounced in those patients with higher initial levels of both PPIX and AST, with the authors suggesting this reduction in patients’ lowered overall risk of liver injury. Zinc protoporphyrin (ZnP) also decreased during treatment. ZnP is found in red blood cells when heme production is inhibited by causes such as lack of iron. The authors explained this as an improved utilisation of systemically available iron.
These two findings are important developments, adding to our broader understanding of the mechanisms by which afamelanotide may be able to assist EPP – and perhaps other – patients over the long-term. When, in addition to providing systemic photoprotection, treatment with afamelanotide can be shown to offer hepatoprotection, it would even further reduce the burden of the disease and provide all round protection in this rare metabolic disorder. A second study is under way to confirm these first published findings and following conclusion; we will then assess the most appropriate pathway forward.
These two findings are important developments, adding to
our broader understanding of the mechanisms by which afamelanotide may be able to assist EPP – and perhaps other – patients over the long-term.
The second paper2, from the Dutch EPP expert reference centre, has looked at the impact of afamelanotide treatment on circadian rhythms (a natural, internal process that regulates the sleep-wake cycle) and overall light exposure, matching a cohort of EPP patients undergoing treatment to both untreated patients and a control group. Over the course of the study, it was found that afamelanotide treatment allowed patients to expose themselves to more “white light” in the springtime, resulting in less pain, and which helped normalise patients’ circadian rhythms. As disturbances of circadian rhythm can have a negative impact on mental health, sleep, and physical activity, the normalisation of the circadian rhythm can be seen as contributing to a better overall health status in treated EPP patients.
Clinical Programs
(Dr Pilar Bilbao, Head of Clinical Operations)
An overview of key developments in CLINUVEL’s expanded clinical program is provided below:
Arterial Ischaemic Stroke
Earlier this month we announced completion of patient enrolment in the CUV801 study, the world’s first study of the safety and efficacy of afamelanotide as a treatment for patients experiencing an arterial ischaemic stroke (AIS). Afamelanotide treatment was well tolerated by all six patients, with no adverse drug reactions reported. The study will now be completed, and the results analysed and shared during 2022. The next studies are already being prepared, taking into account the learnings from the clinical observations and learnings from CUV801.
Given an estimated 15 million strokes are suffered worldwide each year, it is safe to assume that most o the population has been affected – directly or indirectly – by stroke. Many stroke patients have permanent brain damage and disability for life, losing their ability to function or care for themselves unassisted, and there is an estimated 40% risk that patients will incur a stroke in the subsequent decade of life (11% within the first year).3 The burden of this acute condition for those patients who do survive is immense, and generally falls to immediate family members who must often fundamentally restructure their entire lives to provide necessary care.
The study will now be completed, and the results analysed and shared during 2022. The next studies are already being prepared, taking into account the learnings from the clinical observations and learnings from CUV801.
Approximately 85% of all strokes are ischaemic strokes, the result of a clot lodged in a brain artery depriving cells of oxygen and nutrients. Due to access to treatment and narrow “therapeutic windows” during which treatment can be administered, a considerable number of these patients (up to 85% in one European study4) are considered ineligible for either the pharmaceutical (thrombolysis) or surgical intervention (thrombectomy) standards of care. Put simply: physicians lack further tools necessary to combat stroke and help patients recover.
It is because of this unmet need, and the evidence supporting the use of afamelanotide in stroke, that we commenced this important and exciting clinical program.
As a first ever study of afamelanotide in stroke patients, safety remains the key focus of CUV801. Here our teams closely monitor key health outcomes for patients before, during, and after treatment to understand the impact that the therapy may have upon their overall health. Adverse reactions – unwanted or undesirable effects which may be attributed to the interventional drug treatment – are an all-essential factor here, since life is at stake. The fact that, to date, we have had no adverse reactions related to afamelanotide treatment in the study gives us much comfort that the program in stroke can continue. We now await the completion of the study by the final patients and formal data analyses to better understand the impact of afamelanotide in this pilot study. One cannot underestimate the focus on safety in the use of our melanocortin(s), which would make a huge difference for the Company for years to come.
To learn more about the science behind CLINUVEL’s arterial ischaemic stroke program, see:
Scientific Communiqué XI: Developing Novel Treatments for Arterial Ischaemic Stroke Patients.
Dr Pilar Bilbao discussing CLINUVEL’s stroke trials on our YouTube channel.
DNA Repair
The DNA Repair program to assess the role of afamelanotide to protect skin and regenerate DNA of the skin damaged from exposure to light is underway. The initial focus is on xeroderma pigmentosum (XP) patients who are 10,000–fold more susceptible to skin cancers, and who suffer a great number of these cancers throughout their lives often leading to surgeries and death. Two XP-C patients have been treated under the CUV156 study. A study of healthy volunteers (CUV151) is planned, as are other studies with XP-C and XP-V variants of the disease. Progress in these studies will be reported throughout 2022.
Vitiligo
Prior to the conclusion of 2021, CLINUVEL announced it will be proceeding to assess the role of SCENESSE® as a monotherapy for the treatment of the skin depigmentation disorder, vitiligo. This culminated a period of liaison with the US Food and Drug Administration (FDA) on the design of the next CUV study. Since narrowband ultra-violet B phototherapy is still not approved by the FDA for the treatment of vitiligo, we agreed to proceed with the assessment of SCENESSE® as a monotherapy for systemic re-pigmentation, focused on patients with darker skin types (IV to VI on the Fitzpatrick scale). We are now working towards the commencement of the pilot study, CUV104.
A study of healthy volunteers (CUV151) is planned, as are other studies with XP-C and XP-V variants of the disease. Progress in these studies will be reported throughout 2022.
Other Programs
CLINUVEL’s focus on other programs continues, including a formulation of afamelanotide for paediatric EPP patients, a study on SCENESSE® as a treatment for variegate porphyria (VP), an indication related to EPP, and a new (sixth) indication, all of which we are planning to progress this year.
Communications and Investor Relations
(Mr Malcolm Bull, Head of Australian Operations & Investor Relations)
The frequency and range of communication to stakeholders will go up another level in 2022. This reflects an increase in the updates likely across the expanded clinical program. The Operations Updates, Strategic Updates and Investor Webinars held in 2021 were well received by the overwhelming majority of stakeholders and will continue to be delivered throughout 2022.
We seek to provide regular information on the progress of the business for the benefit of all stakeholders and specifically, for investors to continue to inform the positive rationale for their investment in this dynamic company.
The plan for the year provides regular presentations at conferences, as outlined below:
| Month | Event |
|---|---|
| January | JP Morgan Healthcare Conference, San Francisco HC Wainwright Bioconnect Investment Conference |
| April | H.C. Wainwright Global Life Sciences Conference |
| May | UBS Global Healthcare Conference |
| June | Jefferies Healthcare Conference |
| September | H.C. Wainwright Global Investment Conference |
| October | Morgans 7th Annual Value in the Vines Investor Conference |
| November | Jefferies London Healthcare Conference |
We have already presented in 2022 to the H.C. Wainwright Bioconnect Conference and participated in the JP Morgan Healthcare Conference, both held earlier this month. Over 550 and 650 companies presented to each conference, respectively. A generally positive outlook for 2022 was gained from the conferences and the feedback on CUV was excellent. The approach conveyed was to “get on with it” to address the coronavirus pandemic and at a company level, to progress their operations and/or clinical programs. This mirrors CLINUVEL’s approach.
In terms of forthcoming updates, we will soon announce cash receipts and expenditures for the December quarter 2021 (the second quarter of financial year 2022) and, in late February, the financial accounts for the half year to December 2021 will be released. There will be a Corporate Briefing on the cash results and an Investor Webinar on the half year results. Announcements on the progress of the clinical program can also be expected. For now, we continue to publish our quarterly results, although the Company is no longer obliged to do so, and I see this as a courtesy to our long-term supporting shareholders.
In terms of forthcoming updates, we will soon announce cash receipts and expenditures for the December quarter 2021 (the second quarter of financial year 2022) and, in late February, the financial accounts for the half year to December 2021 will be released.
Summary and Conclusion
There is no doubt 2022 will be an eventful year for CLINUVEL as we continue to grow and expand into a vertically integrated and diversified biopharmaceutical company. Progress can be expected across the expanded range of clinical programs and in the ongoing growth of commercial operations. The focus of the dedicated CLINUVEL team will be maintained with prudency in the management of risk and costs to ensure the Company progresses its key initiatives with a performance outcome that is as strong as possible in the prevailing environment.
We wish all stakeholders a safe and productive 2022.
Malcolm Bull, Head of Australian Operations & Investor Relations
References:
- Minder, et. al., (2022). Beyond pigmentation: signs of liver protection during afamelanotide treatment in Swiss patients with erythropoietic protoporphyria, an observational study. Therap Ad Rare Dis. 2: 1–17.
- Wensink, et. al., (2022). Objective light exposure measurements and circadian rhythm in patients with erythropoietic protoporphyria: a case-control study, Mol Genet and Metab. ePub 2 January 2022.
- Mohan, et. al., (2011). Risk and Cumulative Risk of Stroke Recurrence. Stroke. 42(5): 1489–94.
- de Sousa et. al., (2019). Access to and delivery of acute ischaemic stroke treatments: A survey of national scientific societies and stroke experts in 44 European countries. Eur Stoke J. 4(1):13–28.
Authorised for ASX release by the Board of Directors of CLINUVEL PHARMACEUTICALS LTD.
Forward-Looking Statements
This release contains forward-looking statements, which reflect the current beliefs and expectations of CLINUVEL’s management. Statements may involve a number of known and unknown risks that could cause our future results, performance or achievements to differ significantly from those expressed or implied by such forward-looking statements. Important factors that could cause or contribute to such differences include risks relating to: our ability to develop and commercialise pharmaceutical products; the COVID-19 pandemic and/or other world, regional or national events affecting the supply chain for a protracted period of time, including our ability to develop, manufacture, market and sell biopharmaceutical products; competition for our products, especially SCENESSE® (afamelanotide 16mg), PRÉNUMBRA® or NEURACTHEL®; our ability to achieve expected safety and efficacy results in a timely manner through our innovative R&D efforts; the effectiveness of our patents and other protections for innovative products, particularly in view of national and regional variations in patent laws; our potential exposure to product liability claims to the extent not covered by insurance; increased government scrutiny in either Australia, the U.S., Europe, Israel, China and Japan of our agreements with third parties and suppliers; our exposure to currency fluctuations and restrictions as well as credit risks; the effects of reforms in healthcare regulation and pharmaceutical pricing and reimbursement; that the Company may incur unexpected delays in the outsourced manufacturing of SCENESSE®, PRÉNUMBRA® or NEURACTHEL® which may lead to it being unable to supply its commercial markets and/or clinical trial programs; any failures to comply with any government payment system (i.e. Medicare) reporting and payment obligations; uncertainties surrounding the legislative and regulatory pathways for the registration and approval of biotechnology and consumer based products; decisions by regulatory authorities regarding approval of our products as well as their decisions regarding label claims; our ability to retain or attract key personnel and managerial talent; the impact of broader change within the pharmaceutical industry and related industries; potential changes to tax liabilities or legislation; environmental risks; and other factors that have been discussed in our 2021 Annual Report. Forward-looking statements speak only as of the date on which they are made, and the Company undertakes no obligation, outside of those required under applicable laws or relevant listing rules of the Australian Securities Exchange, to update or revise any forward-looking statement, whether as a result of new information, future events or otherwise. More information on preliminary and uncertain forecasts and estimates is available on request, whereby it is stated that past performance is not an indicator of future performance.
Contact
Level 11, 535 Bourke St
Melbourne, 3000 Vic,
Australia
+61 3 9660 4900
+61 3 9660 4909